
MetaSite 7
Want to download, evaluate or know more about MetaSite 7?
Please refer to Mass
Analytica
Introduction
Most drugs are metabolized by Cytochrome P450 enzymes, which are primarily found in the liver. These cytochromes, along with about thirty other enzymes, carry out biotransformations known as phase I reactions. Additionally, around twenty other enzymes are responsible for phase II reactions, which generally involve conjugation processes. While cytochromes are involved in most metabolic processes, there is growing interest in understanding the mechanisms of non-CYP metabolic reactions.
In MetaSite 7, there are 23 non-CYP phase I enzymes considered. Besides FMO3 and AOX1, which were already present in MetaSite 6, the list has been expanded to include eight families and their related isoforms. Furthermore, enzymes responsible for the main phase II conjugation reactions have also been added, covering 12 different families and 20 enzymes in total.
In addition to these new enzymes, the computational method used in MetaSite 7 is innovative. Predictions are based not only on the ligand's reactivity-structure relationship but also heavily on a three-dimensional component. This component relies on the 3D structure of the enzyme and the ligand's ability to interact with it, exposing the reactive group and facilitating the reaction mechanism.
MetaSite command-line execution is controlled through script files written in a simple declarative language, allowing for sequential command execution. This language provides flexibility similar to the graphical interface, enabling operations such as opening, saving, and manipulating MetaSite document files. Users can alternate between command-line and GUI execution, such as importing objects and performing calculations via the command line, then browsing results in the GUI. In MetaSite7, these options are expanded to include all non-CYP Phase I enzymes, Phase II enzymes, or a combined CYP -Phase I - Phase II process.
New Features in version 7
A new sophisticated algorithm
The computational algorithm in MetaSite 7.0 is highly sophisticated and uses simulations to replicate how human metabolic enzymes interact with xenobiotic compounds. For the newly introduced enzymes, exposure and reactivity concepts remain crucial for determining whether a molecule is a potential substrate. However, the prediction does not rely on the classical equation used for CYPs. Instead, it uses the concept of reactivity to select reactive atoms for each enzyme based on the molecule's structure and the enzyme's reaction mechanism.
Using the enzyme's 3D structure (either X-ray or modeled), MetaSite 7 evaluates the exposure of the reactive atom to the enzyme's cofactor or catalytic residues in the binding site. The algorithm employs the GRID force field to calculate flexible interaction fields (flexible MIFs) between the active site amino acids and the potential substrate. This flexible coupling generates multiple docking poses, which are evaluated by the GRID force field using functions that sum energy contributions such as van der Waals forces, hydrogen bonding, hydrophobic interactions, electrostatic forces, entropic contributions, and steric effects. The resulting poses are ranked by total energy interaction value, and the best pose is chosen and normalized to produce a probability score.
The method is designed and automated so that the user can easily design or import the 2D structure of the potential substrate and customize the list of enzymes for prediction according to their needs.
New (nonCYP) Phase I enzymes
- Alcohol dehydrogenases
- Aldehyde dehydrogenases
- Aldo-keto reductases
- Carbonyl reductases
- Carboxylesterases
- Hydroxysteroid dehydrogenase
- Monoamine oxidases
- NAD(P)H quinone Oxidoreductase
New Phase II enzymes
- Phosphatases
- Deaminases
- Carboxypeptidases
- Dipeptidases
- Glutathione S-transferases
- Methyltransferases
- Acetyltransferases
- Sulfotransferases
- Glucuronosyltransferase
- Glycine N-acyltransferase
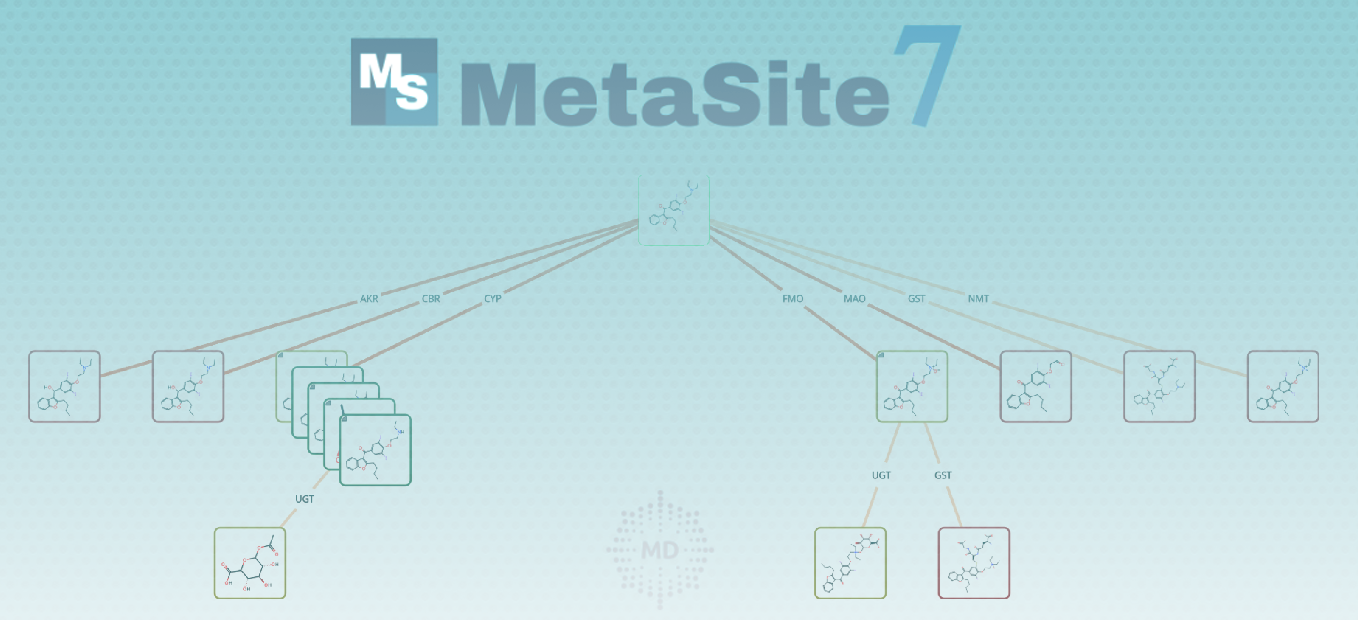
SoM Phase I/Phase II prediction
The result of the SoM prediction for phase I and phase II highlights the predicted sites of metabolism (pSOMs) and provides an overall probability score indicating how likely a given compound is to be a substrate for a specific enzyme. This probability score is derived from the energy of the docking pose, which is determined by exposing the SoM to the enzyme's reactive region (cofactor or catalytic residues). For each predicted SoM, the possible metabolite structure can be determined based on the enzyme's reaction mechanism.
Combined Prediction: Phase I & Phase II
One of the most innovative features is the ability to predict phase I and phase II metabolism in a combined manner. This function predicts phase I metabolism (including both CYP and non-CYP enzymes) for the parent compound and then predicts phase II metabolism for both the parent compound and its phase I metabolites (as selected by the user). This allows for automatic prediction of all first and second-generation metabolites of a molecule, providing a comprehensive view of its potential metabolic pathway in the human body.
Innovative MetID Pathway Analysis
The MetID Pathway Analysis, found in the Metabolites tab, offers a comprehensive overview of predicted metabolic pathways for substrates. It features a graphic panel displaying a network of predicted metabolites and a filter to adjust visibility based on probability. The "ALL" function merges predictions from various enzymes for a full metabolic pathway view, including potential second-generation metabolites. This innovative feature simulates the real metabolic network, providing valuable insights into complex biotransformations
Isoform selectivity function: CYPs stability prediction
(since version 7.1)Cytochromes prediction models available in MetaSite 7 have been added to not only predict the classic SoMs but also to estimate the likelihood of the molecule being metabolized by these enzymes. Each processed molecule is classified as having a LOW, MEDIUM, or HIGH probability of being a substrate based on the predicted probability scores.
REVENG (Reverse Engineering) function: from substrate/metabolite pairs to all possible predicted metabolic paths
(since version 7.2 - CLI only)The REVENG function in the MS7 software offers a groundbreaking solution to this issue. Users can input the chemical structure of a metabolite that has been experimentally identified. The software leverages its advanced algorithms to predcit and return possible enzymatic pathways responsible for the metabolite’s formation. Moreover, REVENG classifies these pathways by assigning probability levels to each biotransformation step. This feature streamlines the process of elucidating metabolic pathways, significantly reducing the effort and resources required, and providing valuable insights for drug development and metabolism studies.