
![]()
FLAP 2.2 & WaterFLAP
Screening, Docking, Water Prediction, Pharmacophore Elucidation, 3D-QSAR
FLAP (Fingerprints for Ligands and Proteins) provides a common reference framework for comparing molecules, using GRID Molecular Interaction Fields (MIFs). The fingerprints are characterised by quadruplets of pharmacophoric features and can be used for ligand-ligand, ligand-receptor, and receptor-receptor comparison. In addition, the quadruplets can be used to align molecules, and a more detailed comparison of the GRID MIF overlap calculated. When the template is a ligand, this enables ligand-based virtual screening and alignment. When the template is a receptor site, this enables structure-based screening and pose prediction. FLAP 2 includes WaterFLAP, a new approach to predicting binding site waters and using them for structure-based design. The waters are scored using two new GRID fields; the CRY field for combined hydrophobicity and lipophilicity, and the ENTR field to estimate the entropic character of a particular water molecule. Combined with the GRID OH2 water enthalpy prediction, waters can therefore be assessed as to their structural, displaceable, or bulk character. The docking engine FLAPdock has been extensively re-parameterized and validated on hundreds of crystal structures, and now includes the ability to use the WaterFLAP waters to guide the docking. GRID is also directly accessible to characterise binding sites in terms of their Molecular Interaction Fields to enable straightforward structure-based design.
Key Features
- Fast ligand-based and structure-based virtual screening using FLAP fingerprints
- Improved virtual screening accuracy using FLAP alignment and GRID MIF scoring
- Optional user-specified pharmacophoric feature constraints
- Ligand-based alignment and structure-based pose prediction using GRID MIFs
- Pharmacophore elucidation and screening
- Automatic pocket detection for structure-based design
- GRID MIF calculation using all standard GRID probes
- Electron density visualisation
- Linear Discriminant Analysis enables the training of target focused scoring functions
- Enrichment plot analysis enables screening approach validation prior to prospective screening
- WaterFLAP water prediction, scoring, and analysis
- FLAPdock docking using GRID MIFs and WaterFLAP waters
- Fuzzy maximal common substructure alignment
- 3D-QSAR analysis
- MoKa integration for automatic protonation and tautomer enumeration and selection (requires FLAP-Suite edition or separate MoKa-Suite license)
- VolSurf+ integration to enable virtual screening that includes pharmacokinetic properties (requires separate VolSurf+ license).
- Parallelisation of virtual screening using MPI for extra high-throughput on Beowolf clusters
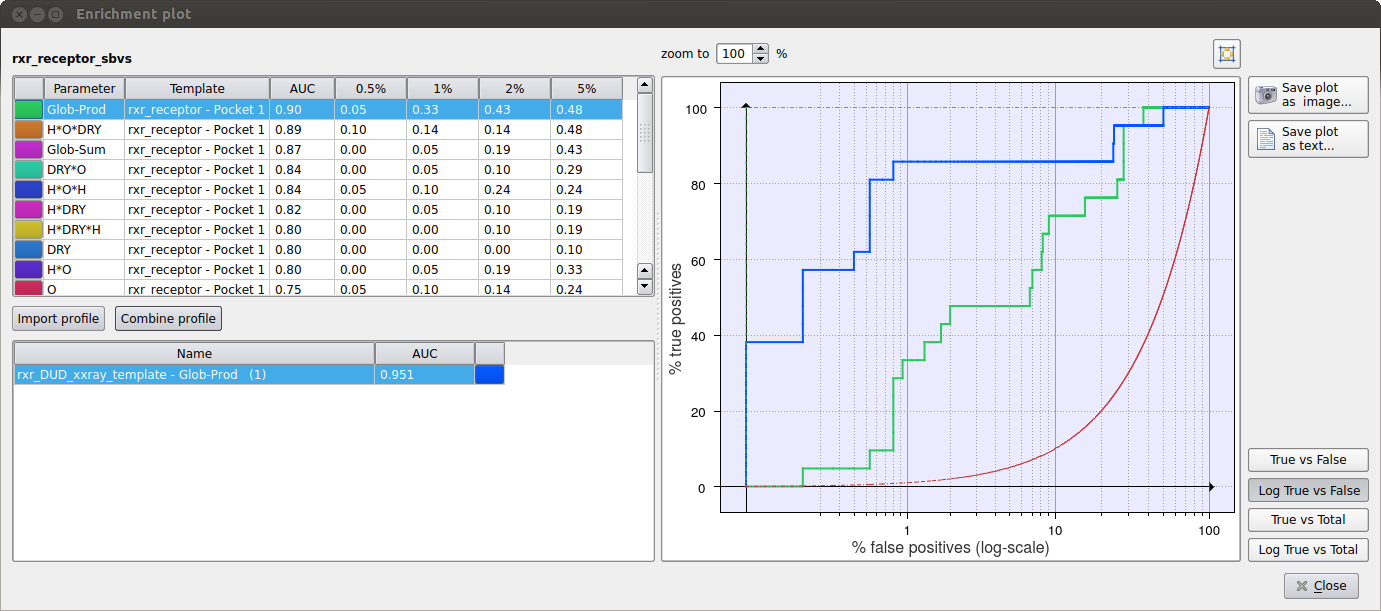
Figure 1. Virtual screening enrichment analysis to select the best scoring function on a set of RXR-alpha inhibitors.
FLAPpharm is a derivative method that uses the FLAP ligand-based alignments to derive template-independent common alignment models for a set of active ligands. From the alignment model, the pharmacophoric interaction fields can be calculated and used for both screening, docking, and increase understanding of SAR. The method has been validated using an extensive pharmacophore validation dataset consisting of 81 targets (PharmBench).
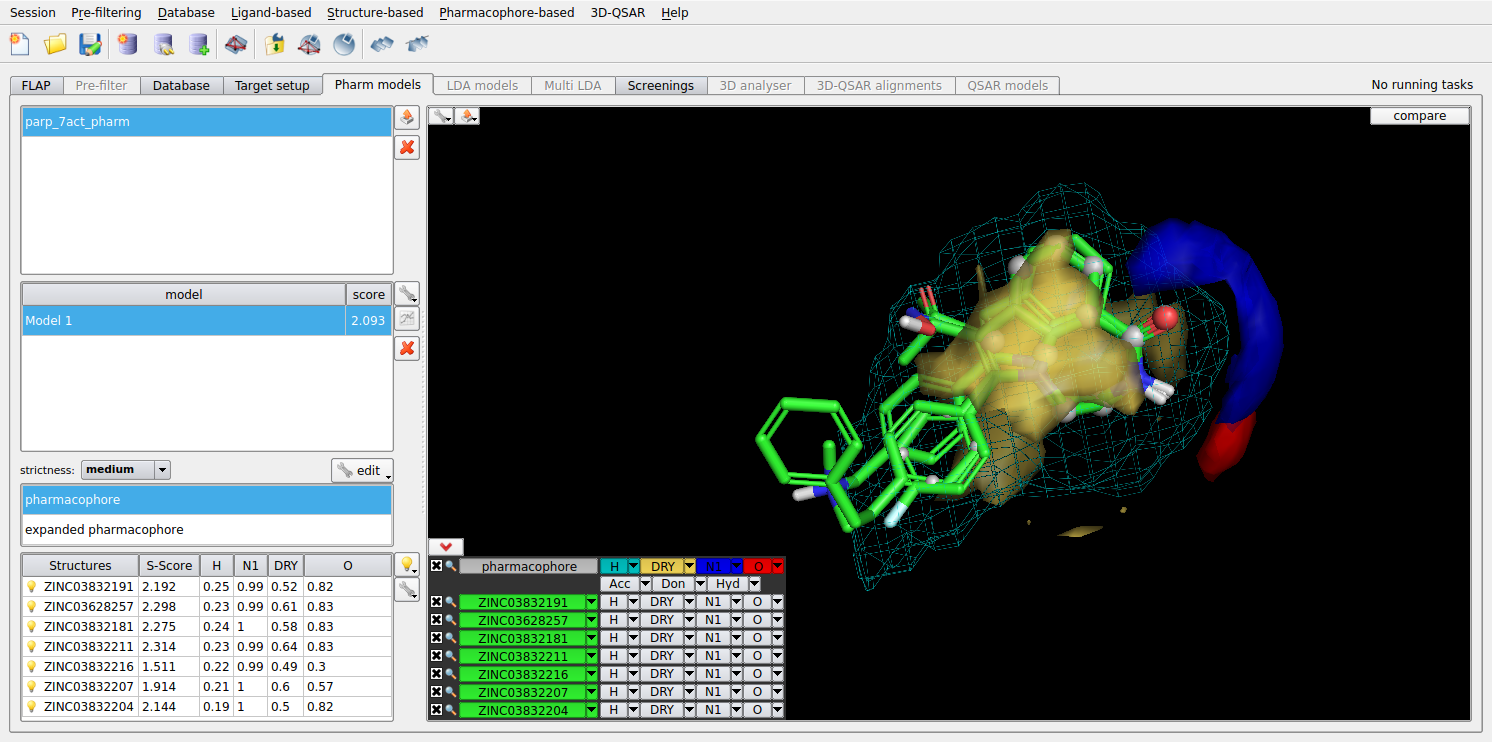
Figure 2. FLAPpharm alignment model and pharmacophoric interaction fields of a set of PARP inhibitors
Given a set of aligned active molecules, FLAP is also able to perform 3D-QSAR by statistical analysis of the GRID MIFs, to enable a detailed examination of an active series and lead optimisation to be performed.
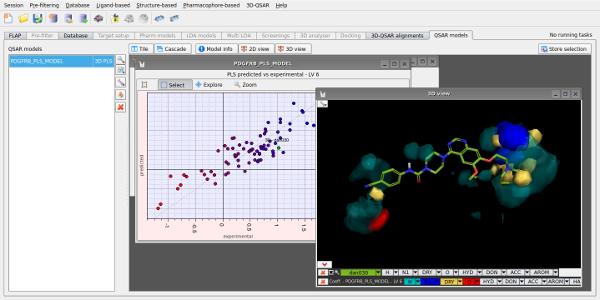
Figure 3. 3D-QSAR analysis for a set of PDGFR inhibitors
Within a binding site, waters are predicted and scored using WaterFLAP. The superimposed ligand is 600-fold less active due to the displacement of a key water within the site, which WaterFLAP highlights as being tightly bound and therefore not displaceable.
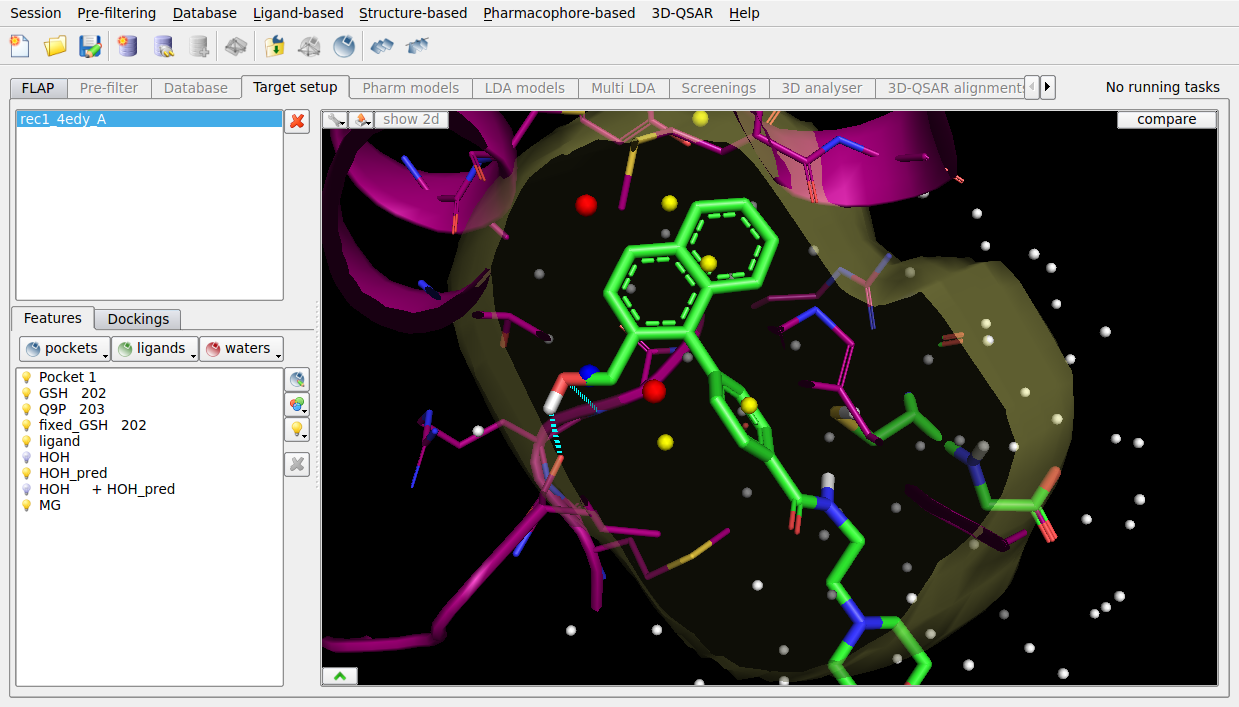
Figure 4. WaterFLAP water prediction for the PGDS binding site
FLAPdock enables flexible fragment-based docking, and can include optional waters from WaterFLAP to guide the docking when water mediate interactions are known to be important. The ligand poses from 4EE0 dock successfully with the WaterFLAP waters, interacting with the key structural binding site water. Docking is not successful without including the key structural water.
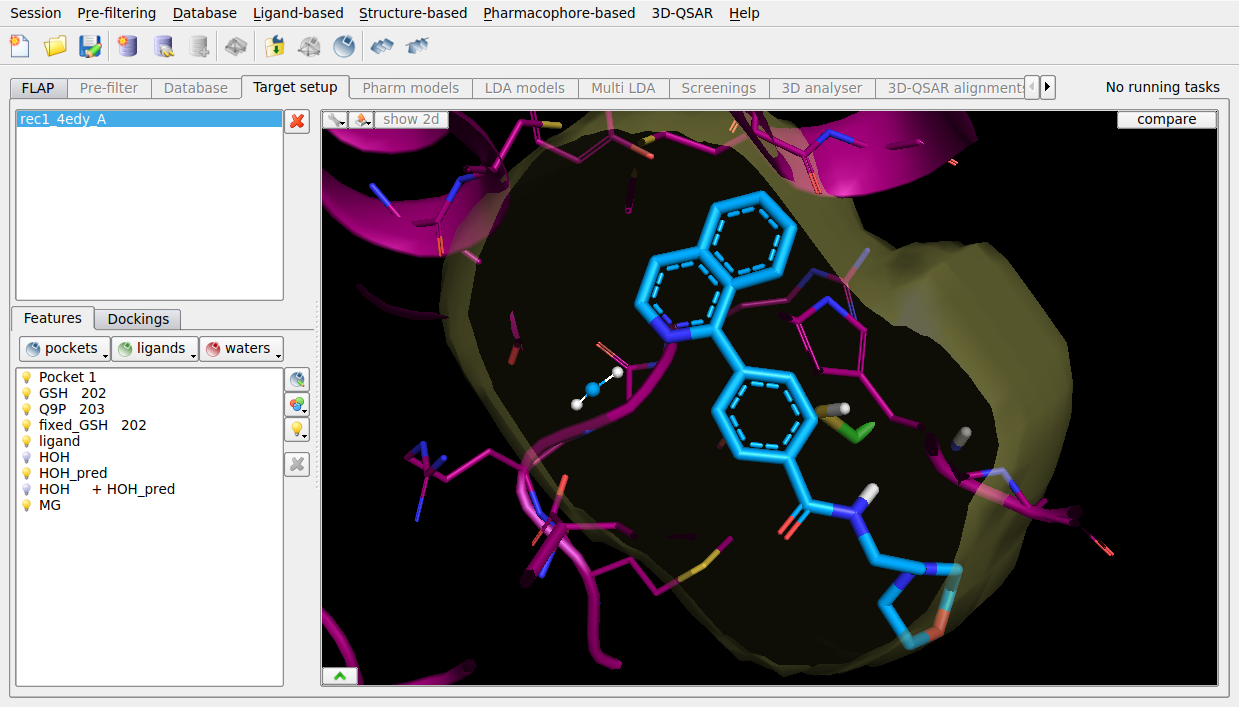
Figure 5. FLAPdock pose prediction of PGDS ligand from 4EE0 using WaterFLAP waters, into the structure of 4EDY.
FLAP also provides a projection of bound ligands and GRID MIFs in the context of the site as a 2D schematic representation to facilitate design and communication.
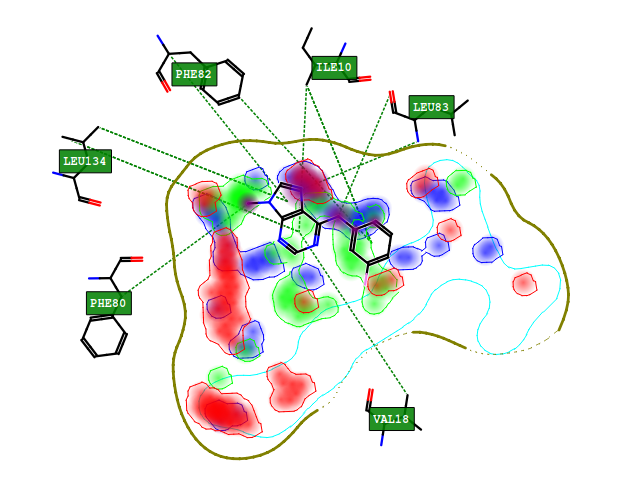
Figure 6. Ligand, FLAPsite pocket, and GRID MIFs from 1CKP projected in 32D mode. Key interactions shown as dotted green lines, the top 5 energetically favourable ligand atoms are highlighted in purple (POSI interactions). GRID MIFs are shown for probes O (red, -5.0 kcal), N1 (blue, -6.5 kcal), DRY (green, -1.0 kcal).
License Options
FLAP Solo provides a license for the FLAP graphical interface and command-line tools for 1 or more CPU
FLAP Suite provides a unlimited (site-wide) access to FLAP GUI, FLAP command-line, and the MoKa pKa tools for tautomer and protomer enumeration and selection.
References
- Tondi F, Cross S, Venturelli A, Costi, M P, Cruciani G, Spyrakis F. Decoding the structural basis for Carbapenem Hydrolysis by class A B -lactamases: Fishing for a Pharmacophore. Current Drug Targets 2016, 17, 983-1005.
- Spyrakis F, Benedetti P, Decherchi S, Rocchia W, Cavalli A, Alcaro S, Ortuso F, Baroni M, Cruciani G. A Pipeline to enhance ligand virtual screening: Integrating Molecular Dynamics and Fingerprints for Ligand and Proteins. J Chem Inf Model, 2015, 55, 2256-2274.
- Siragusa L, Cross S, Baroni M, Goracci L, Cruciani G. BioGPS: Navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins: Structure, Function, and Bioinformatics 2015, 83, 517-532.
- Siragusa L, Spyrakis F, Goracci L, Cross S, Cruciani G. BioGPS: The Music for the Chemo- and Bioinformatics Walzer. Mol. Informatics 2014, 33, 446-453.
- Artese A, Cross S, Costa G, Distinto S, Parrotta L, Alcaro S, Ortuso F, Cruciani G. Molecular interaction fields in drug discovery: recent advances and future perspectives. WIREs Comput Mol Sci 2013, 3:594-613.
- Sirci F, Istyastono E P, Vischer H F, Kooistra A J, Nijmeijer S, Kuijer M, Wijtmans M, Mannhold R, Leurs R, de Esch I J P, de Graaf C, Virtual fragment screening: Discovery of histamine H3 receptor ligands using ligand-based and protein-based molecular fingerprints. J. Chem. Inf. Model. 2012, 52, 3308-3324.
- Cross S, Baroni M, Goracci L, and Cruciani G. GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. J. Chem. Inf. Model. 2012, 52(10):2587-2598.
- Cross S, Ortuso F, Baroni M, Costa G, Distinto S, Moraca F, Alcaro S, and Cruciani G. GRID-Based Three-Dimensional Pharmacophores II: PharmBench, a Benchmark Data Set for Evaluating Pharmacophore Elucidation Methods. J. Chem. Inf. Model. 2012, 52(10):2599-2608.
- Sirci F, Goracci L, Rodriguez D, van Muijlwijk-Koezen J, Gutierrez-de-Teran H, and Mannhold R. Ligand-, structure- and pharmacophore-based molecular fingerprints; a case study on adenosine A1, A2A, A2B, and A3 receptor antagonists. J Comput Aided Mol Des 2012, 26, 1247-1266.
- Brincat JP, Carosati E, Sabatini S, Manfroni G, Fravolini A, Raygada JL, Patel D, Kaatz GW, and Cruciani G. Discovery of Novel Inhibitors of the NorA Multidrug Transporter of Staphylococcus Aureus. J. Med. Chem., 2011, 54 (1), pp 354–365.
- Carosati E, Sforna G, Pippi M, Marverti G, Ligabue A, Guerrieri D, Piras S, Guaitoli G, Luciani R, Costi MP, and Cruciani G. Ligand-based virtual screening and ADME-tox guided approach to identify triazolo-quinoxalines as folate cycle inhibitors. Bioorg Med Chem 2010, 18 (22), pp 7773-7785.
- Cross S, Baroni M, Carosati E, Benedetti P and Clementi S. FLAP: GRID Molecular Interaction Fields in Virtual Screening. Validation using the DUD Data Set. J. Chem. Inf. Model., 2010, 50 (8), pp 1442–1450.
- Cross S, Cruciani G. Grid-derived structure-based 3D pharmacophores and their performance compared to docking. Drug Discovery Today: Technologies. Volume 7, Issue 4, Winter 2010, Pages e213-e219.
- Sciabola S, Stanton RV, Mills JE, Flocco MM, Baroni M, Cruciani G, Perruccio F, Mason JS. High-throughput virtual screening of proteins using GRID molecular interaction fields. J Chem Inf Model. 2010 Jan;50(1):155-69.
- Carosati E, Budriesi R, Ioan P, Ugenti MP, Frosini M, Fusi F, Corda G, Cosimelli B, Spinelli D, Chiarini A, Cruciani G. Discovery of novel and cardioselective diltiazem-like calcium channel blockers via virtual screening. J Med Chem. 2008 Sep 25;51(18):5552-65.
- Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J Chem Inf Model. 2007 Mar-Apr;47(2):279-94.
- Carosati E, Mannhold R, Wahl P, Hansen JB, Fremming T, Zamora I, Cianchetta G, Baroni M. Virtual screening for novel openers of pancreatic K(ATP) channels. J Med Chem. 2007 May 3;50(9):2117-26. Epub 2007 Apr 11.