
MARS
Want to download, evaluate or know more about MARS?
Please refer to Mass
Analytica
![]()
MARS (MetAbolomics ReSearch) is a vendor neutral desktop application software endowed with a Graphical User Interface (GUI) specifically developed for untargeted and semi-targeted LC-MS-based metabolomics and exposomics.
Differently form Lipostar, which was specifically designed for LC-MS based lipidomics with dedicated tools and workflows, MARS provides more general algorithms and investigation tools.
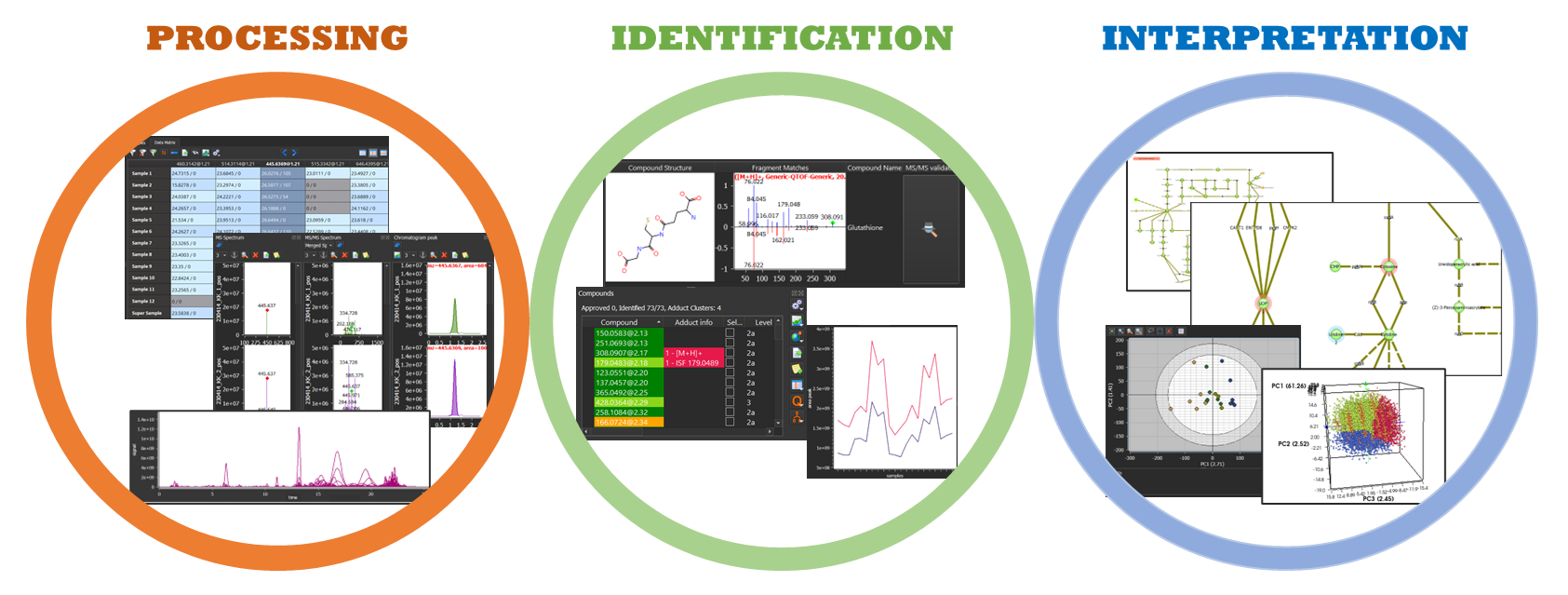
MARS fully covers all the steps required in LC-MS based untargeted and semitargeted metabolomics and exposomics analysis: instrument data conversion and processing, peak detection, statistical analysis, automated MS and/or MS/MS-based metabolite annotation, quantification, and biopathway analysis. Unique features have been developed in the software to improve annotation accuracy, including customizable identification of multiple adducts, automated in-source fragmentation detection, and in-silico MS/MS spectrum validation. Additionally, two MARS databases for exposomics (nitrosamines) and phytomics applications are available upon request.
Features in MARS
Database generation
- The MARS DB Manager module allows to generate customized databases based on internal data as well as automatically import data from The Human Metabolome Database (HMDB), MassBank of North America (MoNA), and Microbial Metabolites Database (MiMe). As already mentioned, two MARS databases for exposomics (nitrosamines) and phytomics applications are available upon request.
Data processing
- Specific data processing algorithm:
- Baseline and noise reduction
- Peak extraction
- Peak smoothing (Statistical Deconvolution Algorithm or Savitzky-Golay)
- Signal-to-noise ratio
- Retention time (RT) correction
- Alignment
- Deisotoping
- Gap-filler (optional algorithm to reduce missing values in the data matrix)
- A new peak detection algorithm for the processing of ion mobility spectrometry (IMS) data (IMS data
are currently supported for Agilent, Waters, and Bruker).
Data matrix refinement
- Several tools for data matrix refinement:
- Filters (e.g., blank subtraction, frequency filter, etc)
- Normalization by metadata (e.g., cell count, volume, weight)
- Normalization by analysis-related data (e.g., standards, total Area, QC, etc)
- Averaging over all replicates
- Merging of positive and negative data matrices
- Adduct clustering
Statistical analysis tool
- MARS provides different analysis to investigate your data:
- Fold-change analysis
- Univariate statistical analysis (e.g., ANOVA)
- Principal Component Analysis (PCA)
- Consensus PCA
- Partial Least Squares regression (PLS)
- Partial Least Squares-Discriminant Analysis (PLS-DA)
- Orthogonal Partial Least Squares (O-PLS)
- Orthogonal Partial Least Squares-Discriminant Analysis (O-PLS-DA)
- Linear Discriminant Analysis (LDA)
Trend Analysis
- An hypothesis-driven approach based on Pearson correlation coefficient or hypothesis-free cluster
analysis (K-means and Bisecting K-means) are supported in MARS to extract trends of interest among
samples.
Metabolite Identification
- A flexible approach for metabolite identification is provided in MARS. It includes:
- A spectral matching approach for species included in the database (RT or CCS values, when available, can be used to improve the annotation accuracy)
- High-throughput approaches to detect other adducts and in-source fragmentations
- A MS/MS validator tool to re-check spectral matching assignation
- Clustering algorithm for adducts and in-source fragments of a same metabolite
- Tool for stable isotope labelling studies
- A score and a level-based classification as index of identification accuracy
- Preliminary search of xenobiotic metabolites
Quantification
- Specific functionalities are provided in MARS for relative and absolute quantification using internal
and/or external standards.
Pathway Analysis
- MARS includes a collection of 20 metabolic pathways obtained by integrating data from different
reference sources (KEGG metabolic network and PathBank linked to HMDB) and literature. The
software also supports the projection of the identification results on metabolic pathways for
functional analysis. The metabolics pathways available in MARS are:
- AAA biosynthesis
- Alanine aspartate and glutamate metabolism
- Arginine and proline metabolism
- Arginine biosynthesis
- Cysteine and methionine metabolism
- Glycolysis and gluconeogenesis
- GSH metabolism
- Histidine metabolism
- Lysine biosynthesis
- Lysine degradation
- N-glycan biosynthesis
- Pentose phosphate pathway
- Phenylalanine metabolism
- Purine pathway
- Pyrimidine metabolism
- TCA cycle
- Tryptophan metabolism
- Tyrosine metabolism
- Valine, leucine and isoleucine biosynthesis
- Valine, leucine and isoleucine degradation
Data support
- MARS supports the import of LC-MS and LC-MS/MS data from the following mass-spec vendors:
- Agilent(*.d): AutoMS and full scan at multiple energies of collision (All Ions).
- Waters(*.raw): MSe, HDMSe, DDA, and MSMS, SONAR.
- Thermo(*.RAW): Ion-Trap and Orbitrap, Exactive, Q-Exactive, DDA and AIF.
- Sciex(*.wiff): SWATH and IDA.
- Bruker(*.d): QTof, FT-ICR, TIMS-TOF data dependent scan.
- Shimadzu(*.lcd): QTof.
- Ion mobility spectrometry (IMS) data are supported for Agilent(*.d), Waters(*.raw), and Bruker(*.d).
- Agilent(*.d), Waters(*.raw), and Shimadzu(*.lcd) files can be directly imported.
- Thermo(*.RAW), Bruker(*.d), and Sciex(*.swiff) files require the use of a converter downloadable from the instrument site.
Requirements
Thermo requirements:
- MSFileReader 3.1 SP3
- MSFileReader 3.1 SP4
Bruker requirements:
- CompassXtract package
Sciex requirements:
- MMS+Wiff+Access+Patch+2-win64.exe
Additional libraries required are listed in the software manual
System requirement and installation
MARS can be installed only on a 64bit Windows operating system.
A detailed installation manual is available
Training
- 12 tutorials are provided.
Publications
- [1] MARS: A Multipurpose Software for Untargeted LC-MS-Based Metabolomics and Exposomics
Laura Goracci, Paolo Tiberi, Stefano Di Bona, Stefano Bonciarelli, Giovanna Ilaria Passeri, Marta Piroddi, Simone Moretti, Claudia Volpi, Ismael Zamora, and Gabriele Cruciani
Analytical Chemistry. 2024, 96, 4, 1468-1477