
MoKa
Accurate pKa prediction and automatic structure modification is critical for many computational chemistry methods which are strongly dependent on the tautomerization and protonation state of the structures, including docking, binding affinity estimation, QSAR and ADME modelling, and metabolism prediction.
MoKa implements a novel approach [1] for in-silico computation of pKa values; trained using a very diverse set of more than 25000 pKa values, it provides accurate and fast calculations using an algorithm based on descriptors derived from GRID molecular interaction fields.
Key Features:
- accurate: prediction error of 0.4 log units (0.7 for novel structures)
- fast: more than one million predictions per hour*
- automatic: output top N most abundant species at a designated pH
- versatile: predicts pKa, logP, logD, logS0, and other properties
- self-trainable: incorporate additional experimental data to improve accuracy
* command line, pKa predictions only, no tautomers generation
MoKa 5 new features and enhancements
- Proteolysis Targeting Chimeras (PROTAC) mode, with specific prediction algorithms for their physicochemical properties
- improved lipophilicity (logP/logD) models with curated experimental data
- salting in/out effect for pH/solubility profile
- vastly improved graphical interface with:
- one-panel view for all the predictions and properties
- flexible naming policy for input structures
- multi-threading predictions engine
- self-encapsulated work session data, allows easy resuming previous work and sharing structures with associated predictions
- further refinement of pKa internal model with new experimental data
More info about changes in Moka are available in the manual
Package content
Graphical interface for predictions

- tautomer check
- batch mode for multi-structure files
- integrated structure editor
- cut & paste from ISIS/Draw (Windows version)
System training module
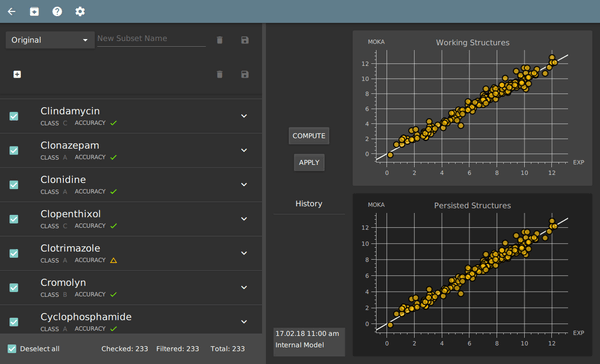
- independent GUI for system training
- automatic assignment of exp. pKa values to ionization sites
- building of customized pKa prediction models to load into MoKa
Command line tools
MoKa command line
- easy integration with existing tools
Generation of ionization states of a compound
- single ionization state mode at user-specified pH value
- multiple ionization states mode at user-specified pH value or pH range
Tautomers enumeration and stability estimation [2]
- batch enumeration of tautomers and estimation of tautomer stability
Availability
MoKa is available for Windows® and Linux® operating systems.
Editions comparison
MoKa comes in two editions: Solo and Suite
MoKa-Solo provides an interactive interface for pKa investigation for 1 or more CPU; Moka-Suite provides unlimited (site-wide) interface access and also command-line utilities for wider enterprise deployment in cheminformatics processes.
This matrix provides an overview of what is provided by the different editions.
| Solo Edition | Suite Edition | |
|---|---|---|
| Graphical interface for predictions | yes | yes |
| Automatic comparisons with experimental data | yes | yes |
| Command line batch predictions | no | yes |
| Generation of ionization states of a compound | no | yes |
| Tautomers enumeration and stability estimation | no | yes |
| System training module | no | yes |